
As informações referentes à doença foram devidamente revisadas pelo Dr. Paulo Victor Sgobbi de Souza e pelo Dr. Wladimir Bocca Vieira de Rezende Pinto (Médicos neurologistas assistentes do Ambulatório de Doenças Neuromusculares da Escola Paulista de Medicina-Universidade Federal de São Paulo – UNIFESP)
______________________________________________________________________
Introdução
A PEH, também conhecida popularmente como paraparesia espástica familiar (PEF), abrange um grupo heterogêneo de distúrbios neurológicos que afetam principalmente os neurônios motores superiores (que transmitem o estímulo motor até a medula espinal), causando fraqueza (paraparesia) e rigidez (espasticidade) nas pernas.
Os primeiros relatos da doença foram realizados em 1880 por Ernst Strumpell (Figura 1). Posteriormente, em 1888, Lorrain fez uma descrição mais detalhada, fazendo com que a doença ficasse conhecida desde então como síndrome de Strumpell-Lorrain.

Figura 1: Ernest Strumpell
A PEH é uma doença neurodegenerativa rara (< 65 em cada 100.000) de origem genética que atinge em torno de 5 em cada 100.000 pessoas, sendo sua epidemiologia frequentemente subestimada por seu difícil diagnóstico e por parte de seus sintomas e sinais ser comum aos evidenciados em outras doenças neurológicas. No Brasil, estima-se que tenhamos 10.000 pessoas com PEH, o que corresponde a 10% da população mundial acometida.
Sintomas
Os sintomas clássicos da doença são a espasticidade e a fraqueza nos membros inferiores. Alguns indivíduos possuem espasticidade e nenhuma fraqueza demonstrável, enquanto outros têm espasticidade e fraqueza na mesma proporção.
A apresentação clínica é fundamentalmente dependente da base genética relacionada ao quadro da PEH em um indivíduo ou família, assim como da interação de tal característica genética com fatores ambientais e medicamentosos.
Os primeiros sintomas geralmente relatados pelos pacientes são o enrijecimento das pernas e/ou dificuldade na marcha. A idade de início destes sintomas é muito variável, podendo comprometer desde a faixa etária pediátrica, incluindo lactentes, até adultos e idosos. Frequentemente, os primeiros sintomas da PEH passam despercebidos e possuem uma progressão lenta, sendo reconhecidos apenas quando provocam maior grau de incapacidade motora e dependência do paciente.
De acordo com o tipo de herança genética, a PEH pode ser classificada em autossômica dominante, autossômica recessiva, ligada ao X ou mitocondrial. Desde 1980, tem sido crescente o número de estudos envolvendo a caracterização genética da PEH, já tendo sido descritos mais de 80 subtipos genéticos distintos de PEH (com milhares de variantes patogênicas relacionadas ao quadro), sendo denominadas sequencialmente de modo numérico de acordo com a ordem de sua descoberta (Tabela 1).
A classificação clínica, baseada na descrição de Anita Harding em 1983, distingue entre formas puras e complexas (ou complicadas) de PEH.
Os principais sintomas da PEH pura são: fraqueza gradual nas pernas, aumento do tônus muscular e rigidez (espasticidade), disfunção urinária e hiperreflexia. Geralmente, a forma pura da doença ocorre mais comumente em casos com padrão de herança autossômico dominante, sendo a SPG4 e a SPG3A os subtipos autossômicos dominantes mais comuns (>40% dos casos). A forma complicada é caracterizada pela ocorrência da paraparesia espástica em associação a sinais e sintomas sistêmicos e neurológicos adicionais, incluindo ataxia cerebelar, parkinsonismo, neuropatia, déficit intelectual, demência, epilepsia, atrofia óptica, surdez neurossensorial, catarata, cardiomiopatia e ictiose. As formas complicadas de SPG mais comuns são a SPG11, SPG15 e SPG7, representando em conjunto mais de 50% das formas autossômicas recessivas de PEH.
A progressão da doença em relação a seus sintomas é muito variável, não obedecendo qualquer tipo de regra, nem mesmo dentro da própria família (variabilidade intrafamiliar), podendo em alguns casos haver estabilização do quadro durante o curso clínico.
A tabela a seguir apresenta os possíveis achados clínicos e de imagem de acordo com os diferentes subtipos de PEH (Fink (2014), Souza et al (2017)).
| Subtipos de PEH | Gene | Achados clínicos e de imagem | |
| SPG1 | L1CAM | RLX; Síndrome de Gareis-Mason; Síndrome de MASA, síndrome de CRASH; hidrocefalia e agenesia do corpo caloso. | |
| SPG2 | PLP1 | RLX; nistagmo, atrofia óptica, hipomielinização severa. | |
| SPG3A | ATL1 |
|
|
| SPG4 | SPAST |
|
|
| SPG5A | CYP7B1 | AR; ataxia cerebelar, ataxia espástica, atrofia óptica. Clique aqui para mais detalhes. | |
| SPG6 | NIPA1 | AD; tremor postural, convulsões. | |
| SPG7 | SPG7 | AR; ataxia cerebelar, oftalmoplegia externa progressiva, atrofia cerebelar | |
| SPG8 | KIAA0196 |
|
|
| SPG9A | ALDH18A1 | AD; paraplegia espástica complicada associada a catarata, refluxo gastroesofágico, neuropatia periférica, acompanhada de forma variável, disartria, ataxia e comprometimento cognitivo. | |
| SPG9B | ALDH18A1 | AR; dismorfismo facial, tremor, déficit intelectual , microcefalia e atrofia do corpo caloso. | |
| SPG10 | KIF5A | AD, parkinsonismo (raramente), ataxia cerebelar, neuropatia, amiotrofia distal, declínio cognitivo. | |
|
SPG11 |
SPG11 |
AR; ataxia cerebelar, parkinsonismo, amiotrofia distal, maculopatia, orelhas de lince, leve alteracao da massa branca. Paraplegia espástica associada de forma variável ao corpo caloso fino, déficit cognitivo, fraqueza nos membros superiores, disartria e nistagmo; pode ter fenótipo semelhante a “síndrome de Kjellin”, com início da infância, paraplegia espástica progressiva acompanhada de retinose pigmentar; 50% do HSP autossômico recessivo é do subtipo SPG11. | |
| SPG12 | RTN2 | AD; PEH clássica pura. | |
| SPG13 | HSPD1 | AD, PEH clássica pura, espasticidade marcada. | |
| SPG14 | 3q27-q28 | AR; neuropatia motora distal, déficit intelectual. | |
| SPG15 | ZFYVE26 | AR; Síndrome de Kjellin, amiotrofia distal, perda auditiva, degeneração da retina, neuropatia, psicose, ataxia cerebelar, parkinsonismo responsivo a levodopa, epilepsia; corpo caloso fino, alterações leve da substância branca. | |
| SPG16 | Xq11 | RLX, afasia do motor, déficit intelectual, hipoplasia maxilar, falanges distalas curtas e grossas | |
| SPG17 | BSCL2 | AD, PEH complicada; fenótipo similar a ELA, paraplegia espástica associada à amiotrofia dos músculos das mãos (Síndrome de Silver). | |
| SPG18 | ERLIN2 | AR; espasticidade severa, convulsões, déficit intelectual, múltiplas contraturas articulares. | |
| SPG19 | 9q | AD; leve neuropatia. | |
| SPG20 | SPG20 | AR; Síndrome de Troyer, dismorfismos múltiplos, amiotrofia distal; ataxia cerebelar (raramente), dispraxia da língua, atrofia cerebelar, leve alteração da substância branca do cérebro. | |
| SPG21 | ACP33 | AR; Síndrome de MAST, declínio cognitivo, neuropatia, corpo caloso fino, atrofia frontotemporal, sinais extrapiramidais e cerebelares. | |
| SPG22 | SLC16A2 | XLSD; ataxia cerebelar, déficit intelectual, distonia, dismorfismos típicos, leucodistrofia hipomielinizante. | |
| SPG23 | 2q24-q32 | AR; PEH complicada, Síndrome de Lison, vitiligo, hiperpigmentação de áreas expostas, lentigos, características faciais, déficit intelectual, neuropatia leve. | |
| SPG24 | 13q14 | AR; PEH pura | |
| SPG25 | 6q23.3-q24.1 | AR; doença do disco intervertebral familiar | |
| SPG26 | B4GALNT1 | AR; déficit intelectual, neuropatia, amiotrofia distal, ataxia cerebelar, nistagmo, distonia, discinesias. | |
| SPG27 | 10q22 | AR; PEH pura | |
| SPG28 | DDHD1 | AR; PEH pura | |
| SPG29 | 1p31.1-p21.1 | AD; perda auditiva, antecipação genética, hérnia de hiato. | |
| SPG30 | KIF1A | AR; neuropatia leve, ataxia leve cerebelar, atrofia cerebelar leve. | |
| SPG31 | REEP1 | AD; amiotrofia distal | |
| SPG32 | 14q12-q21 | AR; déficit intelectual leve | |
| SPG33 | ZFYVE27 | AD; PEH pura | |
| SPG34 | Xq24-q25 | RLX; PEH pura | |
| SPG35 | FA2H | AR; Síndrome de FAHN, declínio cognitivo, ataxia cerebelar, distonia, atrofia óptica, leucodistrofia, acumulação de ferro cerebral nos núcleos da base; estrabismo e convulsões (raramente). | |
| SPG36 | 12q23-q24 | AD; neuropatia. | |
| SPG37 | 8p21.1-q13.3 | AD; PEH pura. | |
| SPG38 | 4p16-p15 | AD; Fenótipo similar a ELA, semelhanças com a Síndrome de Silver. | |
| SPG39 | PNPLA6 | AR; similar a syndrome de Troyer, Amiotrofia distal marcada, atrofia cerebelar e atrofia da medula espinhal. | |
| SPG40 | Unknown | AD; PEH pura, antecipação genética. | |
| SPG41 | 11p14.1-p11.2 | AD; PEH pura. | |
| SPG42 | SLC33A1 | AD; PEH pura. | |
| SPG43 | C19ORF12 | AR; Famílias malianas e brasileiras, amiotrofia severa, contratura múltipla. | |
| SPG44 | GJC2 | AR; ataxia cerebelar, déficit intelectual, perda auditiva, convulsões, espasmos dolorosos, corpo caloso fino, leucodistrofia hipomielinizante. | |
| SPG45/SPG65 | NT5C2 | AR déficit intelectual, atrofia óptica, contraturas articulares, estrabismo, nistagmo, corpo caloso displásico, alterações da substância branca. | |
| SPG 46 | GBA2 | AR; ataxia cerebelar, déficit intelectual, tremor da cabeça, catarata congênita, perda auditiva, testículos pequenos, atrofia cerebelar, corpo caloso fino. | |
| SPG47 | AP4B1 | AR; hipotonia neonatal, déficit intelectual, dismorfismos, risada estereotipada, caráter tímido, protrusão da língua espástica, distonia, convulsões febris de início tardio, corpo fino do calo, alterações da substância branca. | |
| SPG48 | AP5Z1 | AR; PEH pura, hiperintensidades leves da coluna cervical. | |
| SPG49 | TECPR2 | AR; atraso mental, ataxia cerebelar, refluxo gastroesofágico complicado, infecções respiratórias recorrentes, dismorfismos, apneia central recorrente, convulsões, corpo caloso fino, atrofia cerebelar. | |
| SPG50 | AP4M1 | AR; hipotonia neonatal, déficit intelectual, estrabismo, dismorfismos, convulsões de ataque infantil, ventriculomegalia, corpo caloso fino, alterações da substância branca. | |
| SPG51 | AP4E1 | AR; hipotonia neonatal, déficit intelectual, baixa estatura, dismorfismos, caráter tímido, risada estereotipada, convulsões, nistagmo, contratados articulares, leucodistrofia marcada, ventriculomegalia, atrofia cerebelar. | |
| SPG52 | AP4S1 | AR; hipotonia neonatal, déficit intelectual, contraturas articulares, atitude sorridente, caráter tímido, dismorfismos. | |
| SPG53 | VPS37A | AR; déficit intelectual, distonia, pectus carinatum, hipertricose. | |
| SPG54 | DDHD2 | AR; atraso mental, contraturas articulares, dismorfismos, estrabismo, hipoplasia do nervo óptico, corpo caloso fino, alterações da substância branca, picos lipídicos anormais na espectroscopia. | |
| SPG55 | C12ORF65 | AR; perda visual progressiva, atrofia óptica, estrabismo, neuropatia distal, déficit intelectual, dismorfismos faciais leves, corpo calo hipoplásico. | |
| SPG56 | CYP2U1 | AR; neuropatia, distonia, déficit intelectual, alterações da substância branca, corpo caloso fino, calcificação dos núcleos da base. | |
| SPG57 | TFG | AR; atrofia óptica, neuropatia, contraturas. | |
| SPG58 | KIF1C | AR; ataxia cerebelar, hipodontia, déficit intelectual, microcefalia, baixa estatura, clonus fragmentário, coreoatetose, alterações da substância branca. | |
| SPG59 | USP8 | AR; PEH puro. | |
| SPG60 | WDR48 | AR; nistagmo, neuropatia. | |
| SPG61 | ARL6IP1 | AR; neuropatia grave, acropatia mutilante severa. | |
| SPG62 | ERLIN1 | AR; ataxia cerebelar, amiotrofia distal. | |
| SPG63 | AMPD2 | AR; baixa estatura, alterações da substância branca, corpo caloso fino. | |
| SPG64 | ENTPD1 | AR; déficit intelectual, microcefalia, puberdade tardia, alterações da matéria branca leve. | |
| SPG66 | ARSI | AR; neuropatia grave, corpo caloso fino, colpocefalia, hipoplasia cerebelar. | |
| SPG67 | PGAP1 | AR; atraso global do desenvolvimento, tremor das mãos, agenesia do corpo caloso, hipoplasia de vermis cerebelar, hipomielinização. | |
| SPG68 | FLRT1 | AR; amiotrofia leve, nistagmo, atrofia óptica; sem espasticidade marcada. | |
| SPG69 | RAB3GAP2 | AR; déficit intelectual, surdez, catarata. | |
| SPG70 | MARS | AD; déficit intelectual leve, síndrome nefrótica. | |
| SPG71 | ZFR | AR; PEH pura, corpo caloso fino. | |
| SPG72 | REEP2 | AR/AD; tremor postural. | |
| SPG73 | CPT1C | AD; amiotrofia leve. | |
| SPG74 | IBA57 | AR; neuropatia, atrofia óptica. | |
| SPG75 | MAG | AR; Paraplegia espástica complicada, marcha espástica associada à ataxia, disartria, neuropatia periférica, atrofia óptica e comprometimento cognitivo leve a moderado; alteraçao da substância branca. | |
| SPG76 | CAPN1 | Paraplegia espástica complicada começando entre 19 e 39 anos, acompanhada de forma variável por disartria, ataxia, atrofia muscular, neuropatia periférica. | |
| SPG77 | FARS2 | AR; PEH pura. | |
| SPG78 | ATP13A2 | AR, Paraplegia espástica complicada, marcha espástica lentamente progressiva, acompanhada de forma variavél por ataxia, neuropatia periférica, tremor, demência, distúrbio supranuclear do olhar. | |
| IAHSP | ALS2 | AR; fenótipo ascendente, hipersinal piramidal marcado. | |
| SPOAN | KLC2 | AR; Brasil e Egito; nistagmo, atrofia óptica, hiperidrose, reflexo de assalto acústico acentuado, amiotrofia distal, neuropatia; Parkinsonismo; atrofia leve da coluna vertebral. | |
| CPSQ-I | GAD1 | AR; déficit intelectual, convulsões ocasionais, microcefalia, contraturas múltiplas. | |
| Parapare-sia espás-tica com surdez | XL; tremor, catarata, surdez, baixa estatura, hipogonadismo. | ||
| BICD2- HSP | 9q22.31 | AR; amiotrofia |
AD- autosomico dominante; AR – autosomico recessivo; RLX – recessivo ligado ao X;
ELA – esclerose lateral amiotrófica; MASA – atraso mental, afasia, paraparesia espástica, polegares aductos; CRASH – hipoplasia do corpo caloso, retardo, polegares aductos, paraplegia espástica e hidrocefalia; FAHN – neurodegeneração associada à hidroxilase de ácidos gordos.
Diagnóstico
Para se estabelecer o diagnóstico de paraparesia espástica hereditária consideram-se os seguintes fatores:
- sintomas e sinais clínicos em ambas as pernas, como perda de força (paraparesia) e espasticidade;
- exame clinico evidenciando comprometimento do trato córtico-espinal com hiperreflexia, espasticidade e sinal de Babinski;
- histórico familiar positivo (mas não obrigatório);
- exclusão de outras doenças;
- teste genético com resultado definitivo (não obrigatório).
Por ser uma uma doença hereditária, a história clínica familiar é muito importante; relacionando parentes com características semelhantes, idade de início dos sintomas e sua progressão, achados em registros médicos como neuroimagens, exames neurológicos e testes genéticos e moleculares dos familiares. Tais dados auxiliam na exclusão de outras doenças e na confirmação do diagnóstico de PEH. Porém, dentro de uma mesma história familiar, é comum encontrar variabilidade quanto a idade de início, gravidade clínica e evolução da doença; isso deve-se a heterogeneidade clínica e genotípica características da PEH.
Devem ser realizados exames complementares que possam excluir doenças com sinais e sintomas semelhantes (Figura 2), incluindo exames de neuroimagem do encéfalo e das medulas cervical, torácica e lombossacral. Exames complementares oftalmológicos, otorrinolaringológicos e que abranjam distúrbios neurometabólicos podem também ser programados. Podem ainda ser necessários exames de avaliação dos ácidos graxos de cadeia muito longa, testes sorológicos (HTLV-I/II, esquistossomose, HIV), vitamina E, vitamina B12, cobre e ceruloplasmina.
Existem mais de 80 subtipos genéticos de PEH descritos na literatura, sendo importante a confirmação individual e familiar do subtipo relacionado aos sintomas e sinais observados. A possibilidade de confirmar o diagnóstico clínico com o estudo genético permite oferecer a possibilidade de diagnóstico pré-natal e eventualmente em contextos específicos de diagnóstico genético pré-implantacional. A confirmação genética do diagnóstico clínico define o subtipo da doença e expõe a possibilidade de outras complicações sistêmicas e neurológicas e a identificação e o tratamento precoces destas.
Alguns subtipos podem ter sintomas parecidos com outras doenças neurodegenerativas (Figura 2). Importante ressaltar que caso o teste genético confirme um subtipo de PEH, isso não significa que a pessoa afetada irá apresentar todos os sintomas característicos daquele subtipo, assim como pode acontecer de um indivíduo assintomático nunca vir a apresentar nenhum sintoma da PEH mesmo com o teste genético positivo.
Pesquisas que visam tratamentos específicos para diferentes subtipos genéticos estão em andamento, o que aumenta a importância da realização do teste genético.
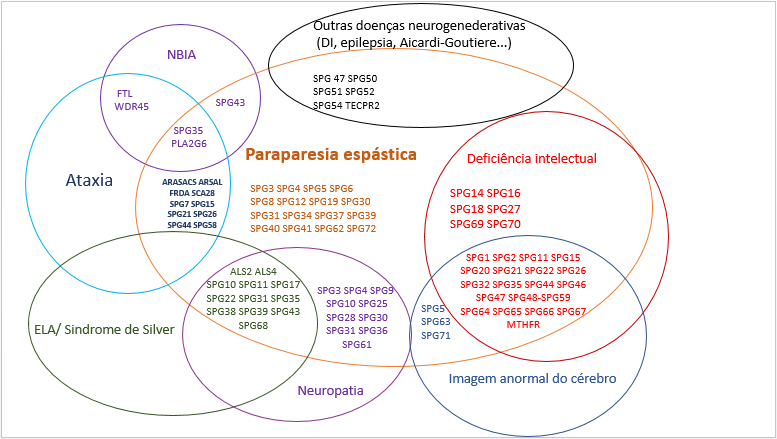
Figura 2. Visão geral da sobreposição clínica e genética entre PEH e outras doenças neurodegenerativas. ELA: esclerose lateral amiotrófica, DI: deficiência intelectual, NBIA: neurodegeneração com acumulação de ferro cerebral.
Tratamento
Embora ainda não haja uma cura para PEH, existem tratamentos que podem ajudar a melhorar a função e o conforto e promover o bem-estar físico e emocional. Como a PEH afeta as pessoas de diferentes maneiras, os programas de tratamento também variam. O recomendado é que o neurologista ou fisiatra desenvolva um programa individualizado baseado no suporte multiprofissional, incluindo ortopedista, fisioterapeuta, terapeuta ocupacional, fonaudiólogo, nutricionista, assistente social ou psicólogo.
Os medicamentos mais utilizados para ajudar a diminuir a espasticidade incluem Baclofeno oral e intratecal, Tizanidina, Diazepam e Clonazepam. Em casos extremos de espasticidade, alguns indivíduos se beneficiam com a toxina botulínica. A Gabapentina, amplamente utilizada para tratar convulsões e dor neuropática, pode também ser útil em alguns casos na redução da espasticidade. Sintomas de fadiga podem ser tratados, conforme indicação médica, com sintomáticos como a Amantadina e a L-Carnitina. A bexiga neurogênica pode ser tratada, depois de adequada avaliação e indicação médica, com drogas anticolinérgicas (como a oxibutinina). A toxina botulínica também pode ser uma alternativa nas injeções locais. Para alguns subtipos genéticos de PEH, podem ser utilizadas outras drogas com objetivo de tratamento específico, como os inibidores da HMG-CoA redutase (estatinas) na SPG5A, e a coenzima Q10 nas formas mitocondriais de SPG ou nas formas com disfunção mitocondrial associada (ex: SPG7).
Cada caso é individual, portanto, para o uso da medicação correta ou para a adoção de outras abordagens terapêuticas para PEH, é fundamental que consulte seu neurologista. Reavaliações clínicas regulares dos pacientes (uma ou duas vezes por ano) são recomendadas para avaliar a progressão e as possíveis complicações.
Referências
Faber et al. (2017). Hereditary spastic paraplegia from 1880 to 2017: an historical review. An historical review. Arq Neuropsiquiatr 75(11): 813-818.
Fink,J.K. Hereditary Spastic Paraplegia Overview. GeneReviews. Disponivel em: < https://www.ncbi.nlm.nih.gov/books/NBK1509/>. Acesso em: 22 de novembro de 2017.
Klebe S, Stevanin G, Depienne C (2015). Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Revue Neurologique, 171(6-7): 505-530.
McDermott CJ, K White, K Bushby, PJ Shaw (2000). Hereditary spastic paraparesis: a review of new developments. J Neurol Neurosurg Psychiatry, 69:150–160.
Polo JM, Calleja J, Combarros O, Berciano J (1993). Hereditary “pure” spastic paraplegia: a study of nine families. Journal of Neurology, Neurosurgery, and Psychiatry, 56:175-181.
Roano et al. (2014). The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology, 42: 174-183.
Souza; P.V.S; Pinto; W.B.V.R; Batistella, G.N. R.; Bortholin, T.; Oliveira, A.S.B. Hereditary Spastic Paraplegia: Clinical and Genetic Hallmarks. Cerebellum (2017) 16: 525. https://doi.org/10.1007/s12311-016-0803-z.
Spastic Paraplegia Foundation: https://sp-foundation.org/
Rodrigues N, Ferreira S, Rodrigues L, Castro A, Barbosa C, Gomes R (2012). Paraplegia espástica familiar tipo 4– antecipação ou variabilidade fenotípica? “Nascer e Crescer” revista do Hospital de Crianças Maria Pia. Vol XXI, n.º 1.